Studies by a growing number of labs have identified neurological health benefits from exposing human volunteers or animal models to light, sound and/or tactile stimulation at the brain’s “gamma” frequency rhythm of 40Hz. In the latest such research at The Picower Institute for Learning and Memory and Alana Down Syndrome Center at MIT, scientists found that 40Hz sensory stimulation improved cognition and circuit connectivity and encouraged the growth of new neurons in mice genetically engineered to model Down syndrome.
Li-Huei Tsai, Picower Professor at MIT and senior author of the new study in PLOS ONE, said that the results are encouraging but also cautioned that much more work is needed to test whether the method, called GENUS (for Gamma Entrainment Using Sensory Stimulation), could provide clinical benefits for people with Down syndrome.
ADSC Director Li-Huei Tsai‘s lab has significantly expanded their Down syndrome (DS) work. New research is looking at brain oscillations and sleep in DS model mice versus neurotypical mice. Continued research on using 40Hz light and sound (GENUS) therapy for several weeks in DS mice has revealed improvements including producing new neurons in the adult brain and reducing brain immune cells. The lab is investigating performance on memory tasks and has also launched a new project testing the benefits of GENUS during pregnancy on development of fetuses with trisomy 21. Finally, the Tsai lab has begun a Phase 1/2 human clinical study with GENUS light and sound devices in individuals with DS. The treatment appears to be safe and well-tolerated with no adverse effects so far.
40Hz light and sound in sleep and neurogenesis
By the age of 40, most individuals with DS present a substantial accumulation of amyloid beta (Aβ) plaques and neurofibrillary tangles (NFT), toxic protein deposits considered to be hallmarks of Alzheimer’s disease. Pioneering studies performed by the Tsai laboratory have shown that sensory stimulation (i.e., light and sound) at the frequency of a key brain rhythm (gamma: 40Hz), a technique called GENUS, enhances gamma rhythms in the brain and can have neuroprotective effects in the context of Alzheimer’s disease. Recent work from the Tsai lab suggests this to be true in mouse models of DS as well. Recent work suggests a key mechanism through which GENUS exerts a neuroprotective effect is by promoting the clearance of waste products through the glymphatic system. The strongest known regulator of glymphatic clearance of waste is sleep. Therefore, a new research project aims to investigate whether GENUS can be performed during sleep and to assess how the potential neuroprotective effects of this treatment compare to the effects of using GENUS during wakefulness. The results of this project could provide a pathway to enhance the beneficial effects of GENUS in the context of Alzheimer’s disease and DS. Moreover, the ability to deliver GENUS during sleep in future clinical trials or interventions would lead to a striking increase in compliance with the treatment and the daily amount of stimulation, particularly for individuals with DS. Taken together, the administration of GENUS during sleep promises a striking increase in treatment efficacy.
To address this aim, Alana Fellow Cristina Blanco-Duque has built a setup that supports continuous (24h/7 days a week) electrophysiology recordings in mice and is equipped to perform GENUS. She also established multi-site in vivo electrophysiology recordings in a mouse model for DS. This setup has already produced several rich datasets with exciting results, including that the DS model mice show fragmented and lighter sleep than their wild-type littermates, and may have hyper-synchronized brain rhythms.
Blanco-Duque and Tsai lab colleagues have also developed a stimulation system that allows the lab to deliver GENUS selectively during sleep or wake. This setup will be used to test whether delivery of the GENUS light and sound treatment is possible during sleep, and how this affects sleep/wake patterns. This system can also be used to study the effects of GENUS on the clearance of toxic waste products during sleep.
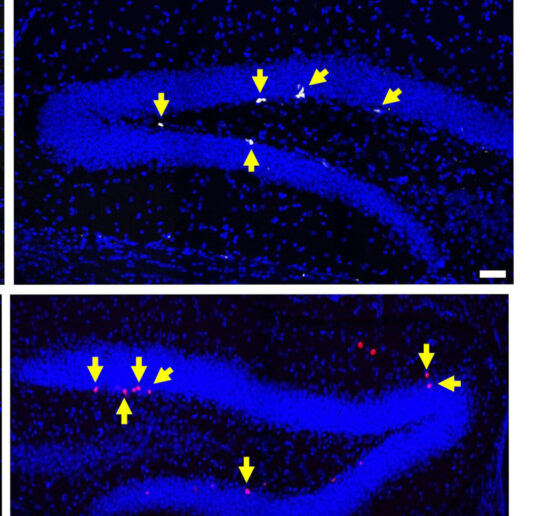
The lab has also found in early results that after three weeks of 40 Hz light and sound (GENUS) stimulation in the mouse model of DS there is a striking benefit of the production of new neurons in the adult brain. This process has been shown previously to be significantly impaired in mouse models of DS, which may contribute to some of the cognitive dysfunction that has been observed. The lab is also investigating the effects of extended GENUS treatment on several behavioral tasks, including spatial short-term memory and anxiety behaviors, as well as markers of inflammation and immune activity in the brain.
Based on preliminary data using single-nucleus RNAseq – a high-resolution method of quantifying gene expression in individual brain cells – the lab hypothesizes that GENUS may alter the expression of several critical DS-related genes that influence the balance between the production of new astrocytes and neurons.
GENUS human clinical studies
The electroencephalogram (EEG) head cap with flat metal discs (electrodes)
Work has started within the Tsai lab to translate the findings in mice into benefits for human subjects with DS. Clinical work began by investigating the safety, compliance, and entrainment of the 40 Hz stimulus. In a recent publication, the Tsai lab found that repeating auditory tones can also be used to entrain gamma oscillations, and in combination with visual stimulation, GENUS can impact brain regions outside of the primary sensory cortex and extend into the hippocampal memory. With combined visual and auditory stimulation at 40Hz, the entrainment of gamma oscillations can be seen across the auditory cortex, visual cortex, hippocampus, and medial prefrontal cortex. Based on these studies, the team developed a non-invasive medical device that will possibly be effective in preventing the progression of AD and other pathology in individuals with DS. Our objective is to determine whether non-invasive sensory stimulation can be used to modulate gamma power and synchronization in individuals with DS as a potential therapeutic to prevent AD and other cognitive troubles in this population.
In a first Phase 1/2 study, the team will treat 30 individuals with DS and age-matched, cognitively typical controls with the GENUS light and sound device while using electroencephalography (EEG) to evaluate induced entrainment and effects on their brain circuitry. Participants are blindly randomized to receive 1-hour of either sham or active 40Hz light and sound stimulation. The team performed cognitive testing before and after the stimulation session. Data so far suggests that our prototype GENUS light and sound device is safe and tolerable in participants with Down syndrome with no significant adverse effects and no evidence of seizure-like activity on EEG during treatment. The lab is continuing to recruit study subjects , with approximately 18 adults with DS already participating in the study.
Prenatal GENUS
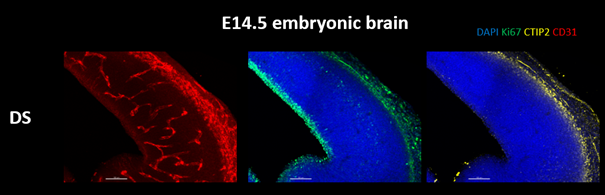
14-day old embryonic mouse brains stained with markers for blood vessels (red) proliferating cells (green) and cortical plate (yellow) in a mouse model of DS
New work in the Tsai lab is finding that the effects of GENUS extend beyond the brain. GENUS stimulation in aged mice changes the number of intestinal immune macrophages and the microbiome composition of the gut. It has been previously shown that immune changes in pregnancy can have large effects on the fetus. Therefore, we started a new project in the lab to understand if GENUS treatment during pregnancy can improve deficiencies found in the brain development of Ts65Dn embryos. The lab is investigating changes in the reduction in brain size and cortical plate thickness observed in Ts65Dn embryos after maternal GENUS treatment. The lab will analyze the maternal plasma, placenta, and brain to understand the possible mechanisms by which GENUS ameliorates the embryonic brain. They also aim to understand if prenatal GENUS treatment can benefit the offspring postnatally in terms of their brain development and function.
In Down syndrome cells, genome-wide disruptions mimic cell aging
Extra chromosome alters conformation and DNA accessibility across the whole genome
The presence of a third copy of chromosome 21 causes all the other chromosomes to squish inward, not unlike when people in a crowded elevator must narrow their stance when one more person squeezes in. The main effects of this are more genetic interactions within each chromosome and less interactions among them. These changes and differences in DNA conformation within the cell nucleus lead to changes in how genes are transcribed and therefore expressed, causing important differences in cell function that affect brain development.
In Down syndrome, the third copy of chromosome 21 causes a reorganization of the 3D configuration of the entire genome in a key cell type of the developing brain, a new study shows. The resulting disruption of gene transcription and cell function are so similar to those seen in cellular aging, or senescence, that the scientists leading the study found they could use anti-senescence drugs to correct them in cell cultures.
The study published in Cell Stem Cell therefore establishes senescence as a potentially targetable mechanism for future treatment of Down syndrome, said Hiruy Meharena, a new assistant professor at the University of California San Diego who led the work as a Senior Alana Fellow in the Alana Down Syndrome Center at MIT.
“There is a cell-type specific genome-wide disruption that is independent of the gene dosage response,” Meharena said. “It’s a very similar phenomenon to what’s observed in senescence. This suggests that excessive senescence in the developing brain induced by the third copy of chromosome 21 could be a key reason for the neurodevelopmental abnormalities seen in Down syndrome.”
The study’s finding that neural progenitor cells (NPCs), which develop into major cells in the brain including neurons, have a senescent character is remarkable and novel, said senior author Li-Huei Tsai, but it is substantiated by the team’s extensive work to elucidate the underlying mechanism of the effects of abnormal chromosome number, or aneupoloidy, within the nucleus of the cells.
“This study illustrates the importance of asking fundamental questions about the underlying mechanisms of neurological disorders,” said Tsai, Picower Professor of Neuroscience, director of the Alana Center, and of The Picower Institute for Learning and Memory at MIT. “We didn’t begin this work expecting to see senescence as a translationally relevant feature of Down syndrome, but the data emerged from asking how the presence of an extra chromosome affects the architecture of all of a cell’s chromosomes during development.”
Genomewide changes
Meharena and co-authors spent years measuring distinctions between human cell cultures that differed only by whether they had a third copy of chromosome 21. Stem cells derived from volunteers were cultured to turn into NPCs. In both the stem cells and the NPCs, the team examined 3D chromosome architecture, several metrics of DNA structure and interaction, gene accessibility and transcription, and gene expression. They also looked at the consequences of the gene expression differences on important functions of these developmental cells, such as how well they proliferated and migrated in 3D brain tissue cultures. Stem cells were not particularly different, but NPCs were substantially affected by the third copy of chromosome 21.
Overall, the picture that emerged in NPCs was that the presence of a third copy causes all the other chromosomes to squish inward, not unlike when people in a crowded elevator must narrow their stance when one more person squeezes in. The main effects of this “chromosomal introversion,” meticulously quantified in the study, are more genetic interactions within each chromosome and less interactions among them. These changes and differences in DNA conformation within the cell nucleus lead to changes in how genes are transcribed and therefore expressed, causing important differences in cell function that affect brain development.
Treated as senescence
For the first couple of years as these data emerged, Meharena said, the full significance of the genomic changes were not apparent, but then he read a paper showing very similar genomic rearrangement and transcriptional alterations in senescent cells.
After validating that the Down syndrome cells indeed bore such a similar signature of transcriptional differences, the team decided to test whether anti-senescence drugs could undo the effects. They tested a combination of two: dasatinib and quercetin. The medications improved not only gene accessibility and transcription, but also the migration and proliferation of cells.
That said, the drugs have very significant side effects—dasatinib is only given to cancer patients when other treatments have not done enough—so they are not appropriate for attempting to intervene in brain development amid Down syndrome, Meharena said. Instead an outcome of the study could be to inspire a search for medications that could have anti-senolytic effects with a safer profile.
Senescence is a stress response of cells. At the same time, years of research by former MIT biology professor Angelika Amon, who co-directed the Alana Center with Tsai, has shown that aneuploidy is a source of considerable stress for cells. A question raised by the new findings, therefore, is whether the senescence-like character of Down syndrome NPCs is indeed the result of an aneuploidy induced stress and if so, exactly what that stress is.
Another implication of the findings is how excessive senescence among brain cells might affect people with Down syndrome later in life. The risk of Alzheimer’s disease is much higher at a substantially earlier age in the Down syndrome population than among people in general. In large part this is believed to be because a key Alzheimer’s risk gene, APP, is on chromosome 21, but the newly identified inclination for senescence may also accelerate Alzheimer’s development.
In addition to Meharena and Tsai, the paper’s other authors are Asaf Marco, Vishnu Dileep, Elana Lockshin, Grace Akatsu, James Mullahoo, Ashley Watson, Tak Ko, Lindsey Guerin, Fatema Abdurrob, Shruti Rengarajan, Malvina Papanastasiou and Jacob Jaffe.
The Alana Foundation, the LuMind Foundation, Burroughs Wellcome Fund, UNCF-Merck and the National Institutes of Health funded the research.
Down syndrome symposium highlights clinical, fundamental progress
Speakers describe studies to address Alzheimer’s disease, sleep apnea and to advance fundamental discoveries
Alana Center co-director Li-Huei Tsai, speaking at a previous center event.
Whether they are working with patients in clinical trials or with chromosomes in cell cultures, scientists and physicians in the Boston area and beyond are testing a wide variety of new ways help people with Down syndrome. At the New England Down Syndrome Symposium, presented by the Alana Down Syndrome Center on Nov. 10, a virtual audience of hundreds of people learned about the research progress of a dozen research teams. The Alana Center at MIT partnered with the Massachusetts Down Syndrome Congress and the LuMind IDSC Foundation to organize the daylong program of online talks.
“I am hopeful that the research being done today will improve medical care and the quality of life of people with Down syndrome,” said Kate Bartlett, a member of the Self-Advocate Advisory Council of the MDSC. “Your work is important for me and my peers. Together we can make a better world for all people to lead active, healthy, fulfilling lives.”
Clinical studies
One of the specific health concerns Bartlett, who is 35, called out in her remarks is that the age of onset for Alzheimer’s disease among people with Down syndrome can be as early as 40. Finding ways to address the community’s elevated risk of Alzheimer’s was one of the four main themes of the symposium, along with new potential therapies for sleep apnea, and fundamental research on developmental biology and on chromosome number and dosage.
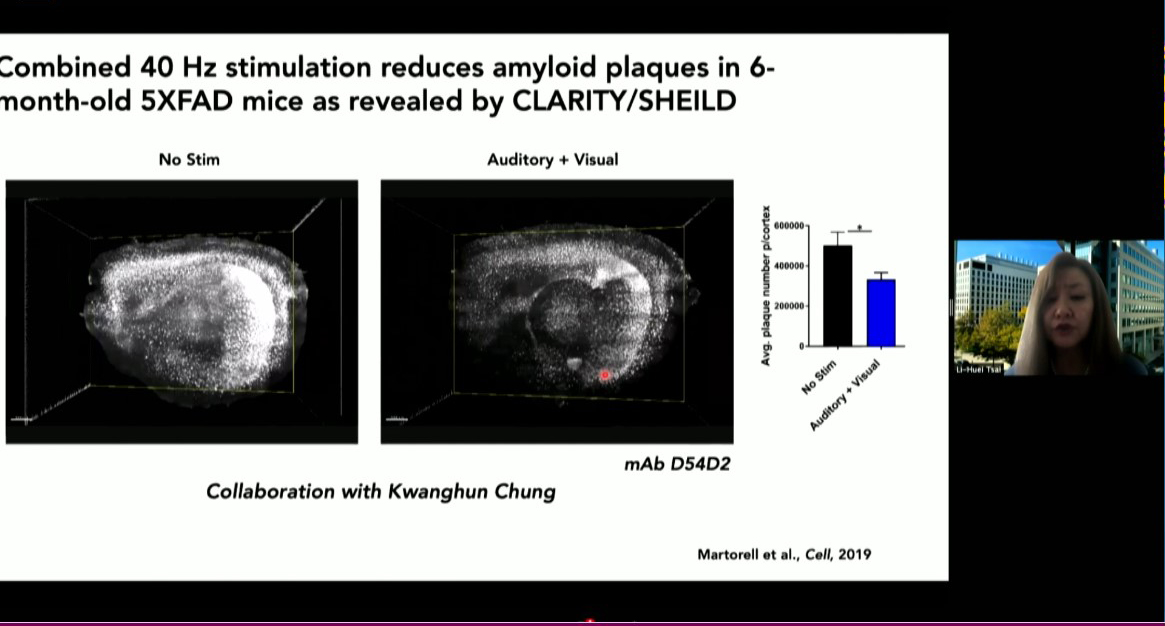
Alana Center co-director Li-Huei Tsai presents research showing that 40Hz light and sound stimulation reduces amyloid plaques, a hallmark of Alzheimer’s pathology. The research team hopes this can help people with Down syndrome, who have an elevated risk of the disease.
MIT is poised to launch a clinical study of a potential Alzheimer’s therapy among people with Down syndrome, said Alana center co-director Li-Huei Tsai, Picower Professor of Neuroscience at MIT. About five years ago her lab discovered that in Alzheimer’s brain wave power and connectivity at a specific frequency, 40Hz, is notably lessened. They discovered that by exposing lab mice to light flickering and sound buzzing at 40Hz they could restore the rhythm, leading to many benefits including improved learning and memory, reduced neuron death and reductions in the level of toxic tau and amyloid proteins considered hallmarks of Alzheimer’s pathology.
More recently the team has begun clinical studies of the potential therapy, called Gamma ENtrainment Using Sensory Stimuli (GENUS), in humans to test its safety and efficacy in healthy people and in people with Alzheimer’s. Picower Clinical Fellow Diane Chan, the neurologist leading the human studies, said that so far the data indicate that exposure to 40Hz light and sound is safe and may be contributing to improved sleep and a preservation of brain volume in patients with mild Alzheimer’s disease. As soon as conditions related to the Covid-19 pandemic allow, she said, the team will invite people with Down syndrome to enroll in a study to test safety, tolerability and efficacy specifically for them.
Two speakers from Massachusetts General Hospital tackled the important related issue of diagnosing and tracking the progression of Alzheimer’s specifically in people with Down syndrome. Stephanie Santoro, a clinical geneticist with the hospital’s Down syndrome program, described the nationwide LIFE-DSR study, which counts Bartlett among its participants. The study has rigorously developed a suite of assessments to track changes in cognition, behavior, function and health in 270 adults of various ages with Down syndrome over a course of more than 30 months, Santoro said. The results will offer doctors and patients new insights into how aging and Alzheimer’s affect life over time, which may help in screening for Alzheimer’s risk.
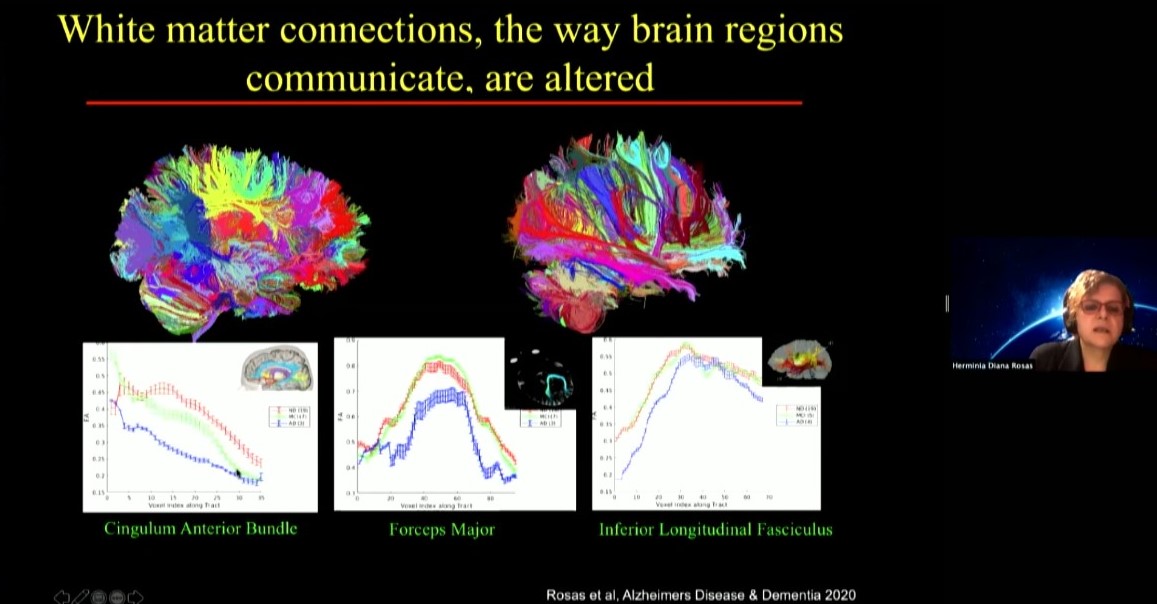
Diana Rosas, an MGH neurologist, is one of the researchers helping to run the LIFE-DSR study. In her talk she focused on another study, the National Institute of Health’s ABC-DS study, in which she is developing biomarkers that may indicate the onset of mild cognitive impairment and Alzheimer’s in Down syndrome including data from brain scans, molecule and protein levels measured in blood, and genetic screens. She noted that these markers seeking to track changes over time need to be specific for Down syndrome patients, for instance because they have characteristic differences in brain anatomy compared to people who don’t have the condition.
MGH neurologist Diana Rosas recently published a study showing differences in brain connectivity among Down syndrome patients with (right) and without Alzheimer’s disease (left).
Another challenge that can hinder learning, memory and cognition in people with Down syndrome is loss of sleep due to breathing trouble. Two symposium speakers discussed new approaches to treating the problem, called sleep apnea, which is very common in people with Down syndrome because of characteristics such as decreased muscle tone, differences in facial anatomy, and larger tongue size. Daniel Combs, a pediatrician at the University of Arizona, described a trial he recently began to test a combination of drugs to treat sleep apnea in children with Down syndrome. The medicines he’s testing have been studied for sleep apnea in non-Down syndrome adults and appear to be helping by increasing airway muscle tone, he said.
Massachusetts Eye and Ear Infirmary otolayrngologist Christopher Hartnick, meanwhile, discussed a surgical approach he is testing for difficult sleep apnea cases. Called hypoglossal nerve stimulation, the procedure involves implanting a breathing sensor on a rib that leads to a processor further up the chest. The processor then stimulates electrodes on muscles of the tongue. When the patient is drawing a breath (sensed at the rib) the processor stimulates the tongue muscles to move the tongue out of the way to improve air flow. This approach has been successful in adults with DS and sleep apnea. So far, Hartnick said, 33 children have been implanted and results look promising.
Fundamental research
At the same time that all of these clinical trials have been progressing, other researchers have been working in the lab to advance more fundamental understanding of the biology going on in cells of people with Down syndrome, often also called trisomy 21 because it is caused by having a third copy of chromosome 21.
Some researchers have forged ahead by working to develop better mouse models of Down syndrome that can more closely reproduce the biology of the condition in the lab. Tarik Haydar of the Center for Neuroscience Research at Children’s National Hospital in Washington DC described his lab’s recent study showing how variations in a predominant mouse model called Ts65dn have led to differing and sometimes contradictory research conclusions that need to be recognized and accounted for.
While important nuances about the Ts65dn model are becoming better understood, Elizabeth Fisher of University College London shared that new mouse models are emerging. In mice the genes that are on human chromosome 21 are spread out over three chromosomes. That has given researchers the challenge of engineering mice to express genes in the same way that people with Down syndrome do. Fisher’s lab has led advances in doing so, and she reported that recently another group managed to directly inserting human chromosome 21 into mice to develop a new model, the TcMAC21 mouse.
Mouse models are crucial because they are whole living organisms that can demonstrate how health and behavior change with an extra chromosome. But another way to model Down syndrome in the lab is by engineering human cell cultures from cells taken from patients. Skin cells, for instance, can be turned into stem cells, which in turn can grow into neurons or heart cells.
MIT biology Professor Laurie Boyer, for instance, has begun studying gene expression in heart muscle cells derived from Down syndrome (DS) persons. The development of the heart is a very intricate and sensitive process and faulty regulation leads to congenital heart defects (CHD). Her goal is to learn how an extra copy of chromosome 21 in DS contributes to the high incidence of CHD that will hopefully fuel potential new therapies for these heart defects.
In other experiments with patient-derived cells—in this case, neurons—Lindy Barrett of the Broad Institute of MIT and Harvard is finding intriguing overlaps between Down syndrome and a form of autism called Fragile X syndrome. Her lab is finding that the protein missing in Fragile X, called FMRP, normally regulates some genes that are also expressed too much in Down syndrome. The findings, she said, raise the question of whether manipulating levels of FMRP could help Down syndrome patients.
While individual genes, or groups of them, could present new targets for therapies, another goal of the field remains finding a way to repress the activity of the third chromosome 21 as a whole. Speakers Jeanine Lee and Mitzi Kuroda, each of Harvard Medical School, described mechanisms by which various organisms, including humans, naturally suppress or enhance whole-chromosome activity. Females have two X chromosomes but males have an X and a Y. To remedy that imbalance, insects like fruit flies doubly express the X chromosome in males but mammals, like people, suppress or “silence” the activity of one of the X chromosomes in females.
This unusual degree of whole-chromosome up- or down-regulation offers intriguing scientific opportunities. Lee discussed how she hopes to address an autism-like disorder called Rett Syndrome in which girls develop abnormally because a mutant copy of the gene MeCP2 happens to be on one X chromosome they express. Her strategy is to selectively subvert X-chromosome silencing to express the healthy copy of MeCP2 on the inactivated X chromosome. Meanwhile speaker Stefan Pinter of the University of Connecticut discussed how his lab is using the X-chromosome’s silencing machinery to silence the extra chromosome 21 in Down syndrome. Pinter said that by silencing the third copy in developing brain cells in the lab his research group is developing a dynamic model for lab studies in which they can now control chromosome 21 dosage in developing brain cells.
In wrapping up the symposium, Alana Center faculty member Ed Boyden, Y. Eva Tan Professor of Neurotechnology at MIT, said the day provided many individual examples of progress that taken together are even more encouraging.
“Today we have seen many individual examples of research and advocacy from which we can draw inspiration and hope,” he said. “But there is another source of those same feelings as well: the way this community came together today to share and to learn from each other. Even if our interaction was virtual, the growth in our understanding and our interconnection was real.”
A path for addressing Alzheimer’s blood-brain barrier impairment
Brain's "keep out" system is compromised in Alzheimer's disease
Detail from a painting by co-author Leyla Akay inspired by the paper.
By developing a lab-engineered model of the human blood-brain barrier (BBB), neuroscientists at MIT’s Alana Down Syndrome Center have discovered how the most common Alzheimer’s disease risk gene causes amyloid protein plaques to disrupt the brain’s vasculature and showed they could prevent the damage with medications already approved for human use.
About 25 percent of people have the APOE4 variant of the APOE gene, which puts them at substantially greater risk for Alzheimer’s disease. Almost everyone with Alzheimer’s, and even some elderly people without, suffer from cerebral amyloid angiopathy (CAA), a condition in which amyloid protein deposits on blood vessel walls impairs the ability of the BBB to properly transport nutrients, clear out waste and prevent the invasion of pathogens and unwanted substances.
In the new study, published June 8 in Nature Medicine, the researchers pinpointed the specific vascular cell type (pericytes) and molecular pathway (calcineurin/NFAT) through which the APOE4 variant promotes CAA pathology.
The research indicates that in people with the APOE4 variant, pericytes in their vessels churn out too much APOE protein, explained senior author Li-Huei Tsai, Picower Directory & Professor of Neuroscience and co-Director of the Alana Down Syndrome Center. APOE causes amyloid proteins, which are more abundant in Alzheimer’s disease, to clump together. Meanwhile, the diseased pericytes’ increased activation of the calcineurin/NFAT molecular pathway appears to encourage the elevated APOE expression.
There are already drugs that suppress the pathway. Currently they are used to subdue the immune system after a transplant. When the researchers administered some of those drugs, including cyclosporine A and FK506, to the lab-grown BBBs with the APOE4 variant, they accumulated much less amyloid than untreated ones did.
“We identify that there is a specific genetic pathway that is expressed differently in a population that is susceptible to Alzheimer’s disease,” said study lead author Joel Blanchard, a postdoc in Tsai’s lab. “By identifying this we could identify drugs that change this pathway back to a non-diseased state and correct this outcome that’s associated with Alzheimer’s.”
Building barriers
To investigate the connection between Alzheimer’s, the APOE4 variant and CAA, Blanchard, Tsai and co-authors coaxed human induced pluripotent stem cells to become the three types of cells that make up the BBB: brain endothelial cells, astrocytes and pericytes. Pericytes were modeled by mural cells that they tested extensively to ensure they exhibited pericyte-like properties and gene expression.
Grown for two weeks within a three-dimensional hydrogel scaffold, the BBB model cells assembled into vessels that exhibited natural BBB properties, including low permeability to molecules and expression of the same key genes, proteins and molecular pumps as natural BBBs. When immersed in culture media high in amyloid proteins, mimicking conditions in Alzheimer’s disease brains, the lab-grown BBB models exhibited the same kind of amyloid accumulation seen in human disease.
With a model BBB established, they then sought to test the difference APOE4 makes. They showed by several measures that APOE4-carrying BBB models accumulated more amyloid from culture media than those carrying APOE3, the more typical and healthy variant.
To pinpoint how APOE4 makes that difference, they engineered eight different versions covering all the possible combinations of the three cell types having either APOE3 or APOE4. When exposed these month-old models to amyloid-rich media, only versions with APOE4 pericyte-like mural cells showed excessive accumulation of amyloid proteins. Replacing APOE4 mural cells with APOE3-carrying ones reduced amyloid deposition. These results put blame for CAA-like pathology squarely on pericytes.
To further validate the clinical relevance of these findings, the team also looked at APOE expression in samples of human brain vasculature in the prefrontal cortex and the hippocampus, two regions crucially affected in Alzheimer’s disease. Consistent with the team’s lab BBB model, people with APOE4 showed higher expression of the gene in the vasculature, and specifically in pericytes, than people with APOE3.
“That is a salient point of this paper,” said Tsai. “It’s really cool because it stresses the cell-type specific function of APOE.”
A pathway toward treatment?
The next step was to determine how APOE4 becomes so overexpressed by pericytes. The team therefore identified hundreds of transcription factors – proteins that determine how genes are expressed – that were regulated differently between APOE3 and APOE4 pericyte-like mural cells. Then they scoured that list to see which factors specifically impact APOE expression. A set of factors that were upregulated in APOE4 cells stood out: ones that were part of the calcineurin/NFAT pathway. They observed similar upregulation of the pathway in pericytes from human hippocampus samples.
As part of their investigation of whether elevated signaling activity of this pathway caused increased amyloid deposition and CAA, they tested cyclosporine A and FK506 because they tamp pathway activity down. They found that the drugs reduced APOE expression in their pericyte-like mural cells and therefore APOE4-mediated amyloid deposits in the BBB models. They also tested the drugs in APOE4-carrying mice and saw that the medicines reduced APOE expression and amyloid buildup.
Blanchard and Tsai noted that the drugs can have significant side effects, so their findings might not suggest using exactly those drugs to address CAA in patients.
“Instead it points toward the value of understanding the mechanism,” Blanchard said. “It allows one to design a small molecule screen to find more potent drugs that have less off-target effects.”
In addition to Blanchard and Tsai, the paper’s other authors are Michael Bula, Jose Davila-Velderrain, Leyla Akay, Lena Zhu, Alexander Frank, Matheus Victor, Julia Maeve Bonner, Hansruedi Mathys, Yuan-Ta Lin, Tak Ko, David Bennett, Hugh Cam, and Manolis Kellis.
The Robert A. and Renee E. Belfer Family Foundation, the Cure Alzheimer’s Fund, The National Institutes of Health, the Glenn Foundation for Medical Research and the American Federation for Aging Research funded the research.
Written by David Orenstein, Picower Institute for Learning and Memory
Human Models for Neuroscience
Stem cell, genetic technologies enable sophisticated studies of human brain cells and brain "organoids"
An organoid, or "minibrain," labeled to show neurons and glial cells (credit: Kwanghun Chung lab)
Scientists cannot perform many experiments directly on people’s brains, but with new technologies, they can create human brain tissue models and ask important questions via experiments on those. Their findings could help them find new ways to improve brain health.
For decades, neuroscientists seeking to better understand human neurological conditions and develop new therapies have worked with the obvious limitation that a living patient’s brain is not open for investigation or experimentation at the genetic, molecular or cellular scale where many of the brain’s mysteries hide. Nonetheless, they’ve made extraordinary progress with studies in animal models and post-mortem human tissue.
But now neuroscientists are in a whole new era. Three technological breakthroughs over the past 12 years have given them a revolutionary way to study human brain disease: They can create cultures of brain cells derived from individual patients, and even engineer complex, three-dimensional “organoids” that mimic key aspects brain tissue. Scientists are only beginning to harness the potential of these new human cell and tissue models, and Picower Institute labs are helping lead the way. ADSC Director Li-Huei Tsai, and Professors Mriganka Sur and Kwanghun Chung are among a global vanguard that is making and analyzing these new patient-derived testbeds and applying them to study conditions such as Down syndrome, Alzheimer’s disease, Rett syndrome, and Zika virus infection.
While the whole field’s progress with these new capabilities has been rapid, so has been the recognition of their limits. That’s why rather than replacing animal models and other research methods, these new models are becoming integrated as powerful tools to complement broader research programs where findings from multiple methods often enhance each other’s value.
The new wave of breakthroughs began in 2007 when scientists showed how to take a cell from an individual’s body (often a skin cell) and to “reprogram” it to become an “induced pluripotent stem cell” (iPSC) that can then be biochemically guided to become any other cell, like a neuron or a supporting astrocyte or microglia. This development allowed scientists to make the brain cells that they of course would never directly extract from patients. By 2013, scientists began using iPSCs to grow 3D cultures of multiple brain cell types, or “organoids,” that can reproduce key aspects of brain development and intercellular interactions. That same year, scientists demonstrated that a technique called CRISPR/Cas9, could be used for precise genetic editing. Scientists quickly began using that to manipulate the genes in their iPSC-grown cultures and organoids, creating “isogenic pairs” where two otherwise identical stem cells contain a disease-causing or healthy version of a gene.
Tsai and lab members Jay Penney and William Ralvenius summarized and celebrated the significance of these breakthroughs for Alzheimer’s disease research in an August 2019 paper in Molecular Psychiatry.
“In little more than a decade since the advent of human iPSC technologies we have developed the ability to generate all the main brain cell types from pluripotent cells,” they wrote. “Increasingly complex 3D co-culture systems are also emerging that allow us to reconstitute many of the key interactions between brain cells. These technologies have already contributed greatly to our understanding of human development and human disease.”
Sur agreed, “It is perhaps the only way one can study a direct human model – it’s astonishing to be able to grow brain cells from a patient with a disease, derived from that person’s genetic material.”
Abundant applications
Picower labs have embarked on numerous studies using the models. In 2018, Tsai’s lab, led by Yuan-Ta Lin and Jinsoo Seo, published a paper in Neuron in which they used 2D single-cell-type iPSC cultures and 3D mixed-cell-type organoids to study the differences made by two versions of the leading risk gene for Alzheimer’s disease, APOE. People with the APOE4 variant are at much higher risk for the disease than APOE3 carriers but scientists haven’t been sure why. Tsai and Lin’s team used CRISPR/Cas9 to make isogenic pairs of neurons, astrocytes and microglia and spotted several key differences that likely help explain how APOE4 raises disease risk. For instance, APOE4 neurons secreted more potentially harmful amyloid proteins, APOE4 astrocytes showed dysregulated cholesterol metabolism and cleared less amyloid. APOE4 microglia, too, did a poorer job of getting rid of amyloid buildup. Using CRISPR to change APOE4 to APOE3, meanwhile, improved cell activity.
Elsewhere in the the Tsai lab, Joel Blanchard is using iPSCs to model the blood-brain barrier, which stringently filters what comes into and goes out of the brain through the blood stream, so he can see how APOE variants affect that. Researchers are also looking at how myelination – the process of wrapping neurons in a fatty sheath to improve their electrical conductivity – may differ in Alzheimer’s.
As part of the work in the Alana Down Syndrome Center, Tsai’s lab is also using iPSC-based cultures to study the difference that having a third copy of chromosome 21 makes in gene expression in a variety of brain cells and in the development of organoids. At October’s Society for Neuroscience (SfN) annual meeting, the team presented some initial results. Hiruy Meharena observed significant physical changes within chromosomes in various brain cell types with the syndrome’s three copies of chromosome 21 (trisomy) vs. when they have two copies (disomy). These changes, which appear especially prevalent in neural progenitor cells, occur genomewide and result in substantial differences in gene expression that are associated with brain development. Meanwhile, Lin showed how organoids grown from trisomy iPSCs vs. disomy ones have smaller diameters after 30 days of growth and show gene expression differences that may hinder development. Elana Lockshin focused on differences in glial cells, finding, for instance, that trisomy astrocytes don’t migrate during development as readily as in disomy ones.
Elana Lockshin, a member of the lab of Li-Huei Tsai, presents research on how neural support cells, or glia, are affected in trisomy 21.
Sur’s lab has been able to make important findings by using iPSCs as part of its studies of Rett syndrome, an autism-like disorder. In a study published in 2017 in Molecular Psychiatry, a team led by Nikolaos Mellios used isogenic 2D iPSC cultures to find that the disease-causing mutation in the gene MeCP2 led to misregulated forms of RNA that alter a key molecular pathway in early neural and brain development. They also used organoids to show that if they corrected regulation of those RNAs, they could restore healthier development. The study helped to show that Rett syndrome may begin to affect health even earlier than the onset of systems in toddlerhood.
Both the Tsai and Sur labs frequently collaborate with Kwanghun Chung, whose research group is dedicated to developing tools and technologies to help fellow scientists better visualize and quantitatively analyze tissues from the scale of whole human brains down to subcellular components like the synaptic connections between neurons. Chung has not only aided the Sur’ lab’s Rett syndrome studies but has also been working with MIT Health Sciences and Technology Professor Lee Gehrke in his lab’s efforts to quantify differences that may help explain what hinders the growth of organoids – and brains – with Zika infection.
“Organoids were pivotal in helping scientists discover how Zika causes microcephaly,” said Chung lab postdoc Alex Albanese. “We are taking a more in depth look at how Zika is actually modifying the structure and cell populations inside the organoids.”
Innovating past limits
A large part of the work in Chung’s lab, which is led by Albanese and Justin Swaney, aims to overcome some key fundamental limits of organoids. Though they are sometimes called “minibrains,” they really aren’t lab replicas of the real thing. A human brain, while extraordinarily complex, has a well-defined geography. Even though organoids are much simpler – with thousands of cells rather than about 100 billion – they are much more variable in how they turn out. While a real brain will develop highly distinct regions and exactly the right number of ventricles in the right places, organoids will recapitulate only an approximation of, say, cortical structure and may have enough ventricles to look more like Swiss cheese than a brain. Albanese calls this the “snowflake” problem, alluding to how organoids can differ so widely.
So how can they still be valuable models? One has to know how to assess them. Chung’s lab has developed advanced tissue processing technologies that can clarify, preserve, label and enlarge tissues, including organoids, so that properties like physiology and cellular function can be highlighted at all scales. Moreover, his lab has developed an imaging pipeline using light-sheet microscopy that is capable of capturing enormous amounts of data quickly (15 minutes per organoid), so that technicians can thoroughly image many organoids in a day.
Another limitation of organoids is that they aren’t actually that small. A millimeter or two of diameter may seem tiny, but that’s still big enough to present challenges. Traditional microscopes can’t image far enough into them to resolve what’s going on with deeper cells. Chung’s technologies overcome that problem both by clarifying tissue and labeling cells and proteins, but that requires chemically fixing the organoids. Yildirim’s “three photon” microscope technology doesn’t label cells as richly, but it can image all the way through organoids while they are alive and active. That’s how Sur’s lab was able to witness the erratic migration of new neurons. They’ve developed microfluidic multiple-well housings for organoids and devised methods for holding them perfectly steady for long-term imaging while still allowing nutrients and oxygen to circulate around them.
Mriganka Sur’s lab has developed a system for holding an organoid very stably while nutrients circulate around.
Indeed a significant problem associated with large size is that with no blood vessels to carry oxygen and nutrients in or to take waste out, the innermost cells can die. Keeping either the nutrients and oxygen, or the organoid, in motion helps keep them healthier than just growing them in dishes, research has shown. The Tsai lab recently began growing organoids by the thousands using a clever bioreactor device that keeps them spinning in the incubator. Postdoc Ping-Chieh Pao said the system saves space and uses less growth media, while yielding higher quality organoids with more mature neurons and less cell death.
Blood vessels are also integral to brain function. For instance, dysfunction in the blood-brain barrier plays a potentially pivotal role in Alzheimer’s. To build a human model of Alzheimer’s that explicitly accounts for vasculature in sickness and in health, Tsai and Blanchard recently secured a grant from the National Institutes of Health to build a “brain on a chip” that will engineer connections to unify their blood-brain barrier model with a co-culture of many brain cell types. The addition of vasculature and cell types like the oligodendrocytes that produce myelination will simulate a richer degree of Alzheimer’s complexity than human iPSC-based models have to date.
Yet another limitation of organoids has been that they don’t mimic multiple brain regions all that clearly, though they can be chemically coaxed to trend toward one or another. For that reason, organoids don’t provide much of a testbed for understanding the functional significance of inter-regional circuits. Delepine said the Sur lab is interested in experimenting with growing multiple organoids of different regional character on a chip and then nurturing connections between them to see if they can replicate such circuits.
Multiple models together
While Picower researchers and colleagues elsewhere work to make the most of human disease models, there are some limits that seem sure to endure, both for technical and ethical reasons. That means that traditional models, including animals, will remain integral to neuroscience research.
After all, organoids don’t think, behave or remember. They can’t gather any sensory input and do not exhibit consciousness. Lacking all these basic capabilities, they naturally can’t provide any data about how disease affects those vital functions of daily existence. In a recent study where Sur collaborated with MIT biologist Rudolf Jaenisch to test new drugs with the potential to treat Rett syndrome, Jaenisch’s lab screened the compounds on stem-cell derived human neurons, but Sur’s lab tested the most promising ones in mouse models, because that was the only way to see if they improved cognition and behavior.
“The question was, were they valid in a much more functional system,” Sur said.
Mellios’s study, too, drew upon and also utilized findings in Rett model mice.
“Our work in these two studies shows how mouse work can complement human work,” he said.
Especially when fighting disease, researchers will always draw upon whatever systems they can to help them find answers.
New method visualizes groups of neurons as they compute
Fluorescent probe could allow scientists to watch circuits within the brain and link their activity to specific behaviors.
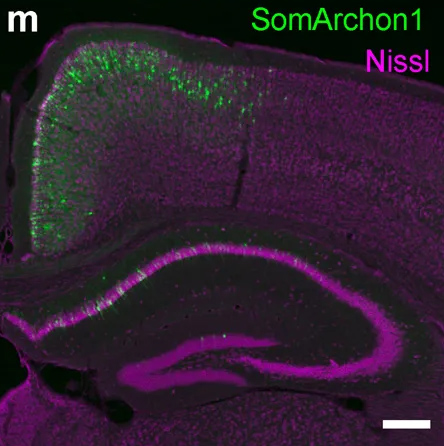
Neurons in a mouse brain are labeled purple. In green, neurons are labeled with a fluorescent probe that reveals electrical activity.
Using a fluorescent probe that lights up when brain cells are electrically active, MIT and Boston University researchers have shown that they can image the activity of many neurons at once, in the brains of mice.
This technique, which can be performed using a simple light microscope, could allow neuroscientists to visualize the activity of circuits within the brain and link them to specific behaviors, says Edward Boyden, the Y. Eva Tan Professor in Neurotechnology and a professor of biological engineering and of brain and cognitive sciences at MIT.
“If you want to study a behavior, or a disease, you need to image the activity of populations of neurons because they work together in a network,” says Boyden, who is also a member of MIT’s McGovern Institute for Brain Research, Media Lab, and Koch Institute for Integrative Cancer Research, and is a member of the Alana Down Syndrome Center.
Using this voltage-sensing molecule, the researchers showed that they could record electrical activity from many more neurons than has been possible with any existing, fully genetically encoded, fluorescent voltage probe.
Boyden and Xue Han, an associate professor of biomedical engineering at Boston University, are the senior authors of the study, which appears in the Oct. 9 online edition of Nature. The lead authors of the paper are MIT postdoc Kiryl Piatkevich, BU graduate student Seth Bensussen, and BU research scientist Hua-an Tseng.
Seeing connections
Neurons compute using rapid electrical impulses, which underlie our thoughts, behavior, and perception of the world. Traditional methods for measuring this electrical activity require inserting an electrode into the brain, a process that is labor-intensive and usually allows researchers to record from only one neuron at a time. Multielectrode arrays allow the monitoring of electrical activity from many neurons at once, but they don’t sample densely enough to get all the neurons within a given volume. Calcium imaging does allow such dense sampling, but it measures calcium, an indirect and slow measure of neural electrical activity.
In 2018, Boyden’s team developed an alternative way to monitor electrical activity by labeling neurons with a fluorescent probe. Using a technique known as directed protein evolution, his group engineered a molecule called Archon1 that can be genetically inserted into neurons, where it becomes embedded in the cell membrane. When a neuron’s electrical activity increases, the molecule becomes brighter, and this fluorescence can be seen with a standard light microscope.
In the 2018 paper, Boyden and his colleagues showed that they could use the molecule to image electrical activity in the brains of transparent worms and zebrafish embryos, and also in mouse brain slices. In the new study, they wanted to try to use it in living, awake mice as they engaged in a specific behavior.
To do that, the researchers had to modify the probe so that it would go to a subregion of the neuron membrane. They found that when the molecule inserts itself throughout the entire cell membrane, the resulting images are blurry because the axons and dendrites that extend from neurons also fluoresce. To overcome that, the researchers attached a small peptide that guides the probe specifically to membranes of the cell bodies of neurons. They called this modified protein SomArchon.
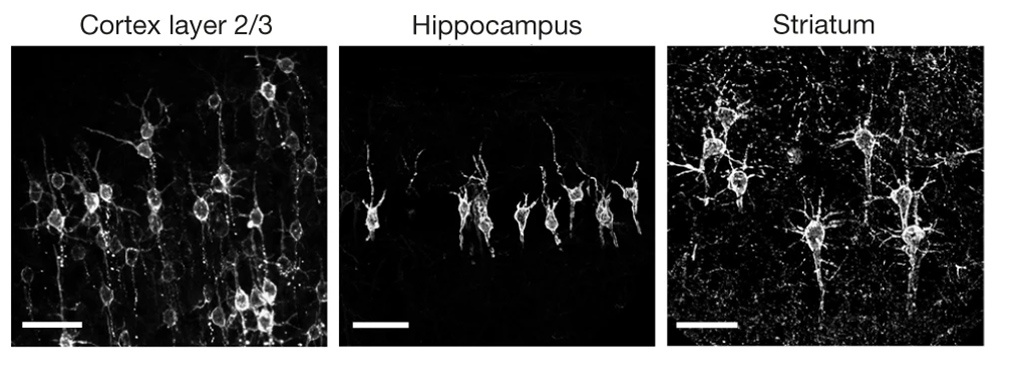
MIT researchers developed a light-sensitive protein that can be embedded into neuron membranes, where it emits a fluorescent signal that indicates how much voltage a particular cell is experiencing. Microscope images of SomArchon-expressing neurons in cortex layer 2/3 (left), hippocampus (middle), and striatum (right)
“With SomArchon, you can see each cell as a distinct sphere,” Boyden says. “Rather than having one cell’s light blurring all its neighbors, each cell can speak by itself loudly and clearly, uncontaminated by its neighbors.”
The researchers used this probe to image activity in a part of the brain called the striatum, which is involved in planning movement, as mice ran on a ball. They were able to monitor activity in several neurons simultaneously and correlate each one’s activity with the mice’s movement. Some neurons’ activity went up when the mice were running, some went down, and others showed no significant change.
“Over the years, my lab has tried many different versions of voltage sensors, and none of them have worked in living mammalian brains until this one,” Han says.
Using this fluorescent probe, the researchers were able to obtain measurements similar to those recorded by an electrical probe, which can pick up activity on a very rapid timescale. This makes the measurements more informative than existing techniques such as imaging calcium, which neuroscientists often use as a proxy for electrical activity.
“We want to record electrical activity on a millisecond timescale,” Han says. “The timescale and activity patterns that we get from calcium imaging are very different. We really don’t know exactly how these calcium changes are related to electrical dynamics.”
With the new voltage sensor, it is also possible to measure very small fluctuations in activity that occur even when a neuron is not firing a spike. This could help neuroscientists study how small fluctuations impact a neuron’s overall behavior, which has previously been very difficult in living brains, Han says.
Mapping circuits
The researchers also showed that this imaging technique can be combined with optogenetics— a technique developed by the Boyden lab and collaborators that allows researchers to turn neurons on and off with light by engineering them to express light-sensitive proteins. In this case, the researchers activated certain neurons with light and then measured the resulting electrical activity in these neurons.
This imaging technology could also be combined with expansion microscopy, a technique that Boyden’s lab developed to expand brain tissue before imaging it, make it easier to see the anatomical connections between neurons in high resolution.
“One of my dream experiments is to image all the activity in a brain, and then use expansion microscopy to find the wiring between those neurons,” Boyden says. “Then can we predict how neural computations emerge from the wiring.”
Such wiring diagrams could allow researchers to pinpoint circuit abnormalities that underlie brain disorders, and may also help researchers to design artificial intelligence that more closely mimics the human brain, Boyden says.
The MIT portion of the research was funded by Edward and Kay Poitras, the National Institutes of Health, including a Director’s Pioneer Award, Charles Hieken, John Doerr, the National Science Foundation, the HHMI-Simons Faculty Scholars Program, the Human Frontier Science Program, and the U.S. Army Research Office.